
Next-generation atomistic simulation
We deliver high-performance, quantum-accurate atomistic simulations based on cutting-edge machine learning methodologies.
We specialize in the Atomic Cluster Expansion (ACE) family of machine learning interatomic potentials, including the Graph Atomic Cluster Expansion (GRACE). Our implementations of these leading methods are carefully engineered for outstanding performance in large-scale atomistic simulations on parallel CPU and GPU architectures.
We generate both custom-made (bespoke) and foundational (universal) atomistic models to drive materials design. The models are trained using advanced machine learning algorithms to reproduce critical material properties accessible by reference electronic structure calculations. Our simulations target a wide range of challenges that arise during the development and application of modern multicomponent materials under diverse environmental conditions.
Use cases

Universal GRACE models are applicable to a broad range of materials and applications
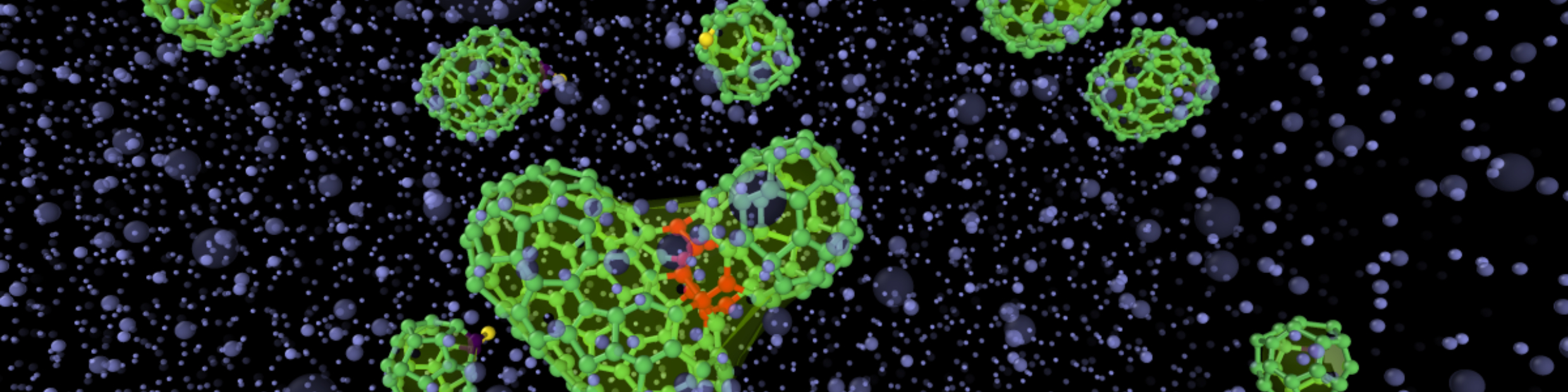
ACE and GRACE enable to follow chemical reactions and their mechanisms
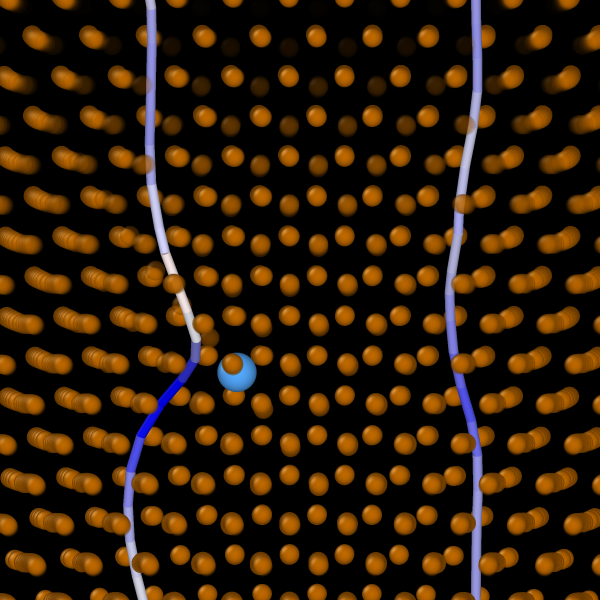
Hydrogen readily interacts with many materials altering their mechanical and functional properties

Large-scale atomistic simulations enable to study microstructural evolution and material failure mechanisms

Magnetic ACE provides a full description of magnetic degrees of freedom and spin-lattice dynamics
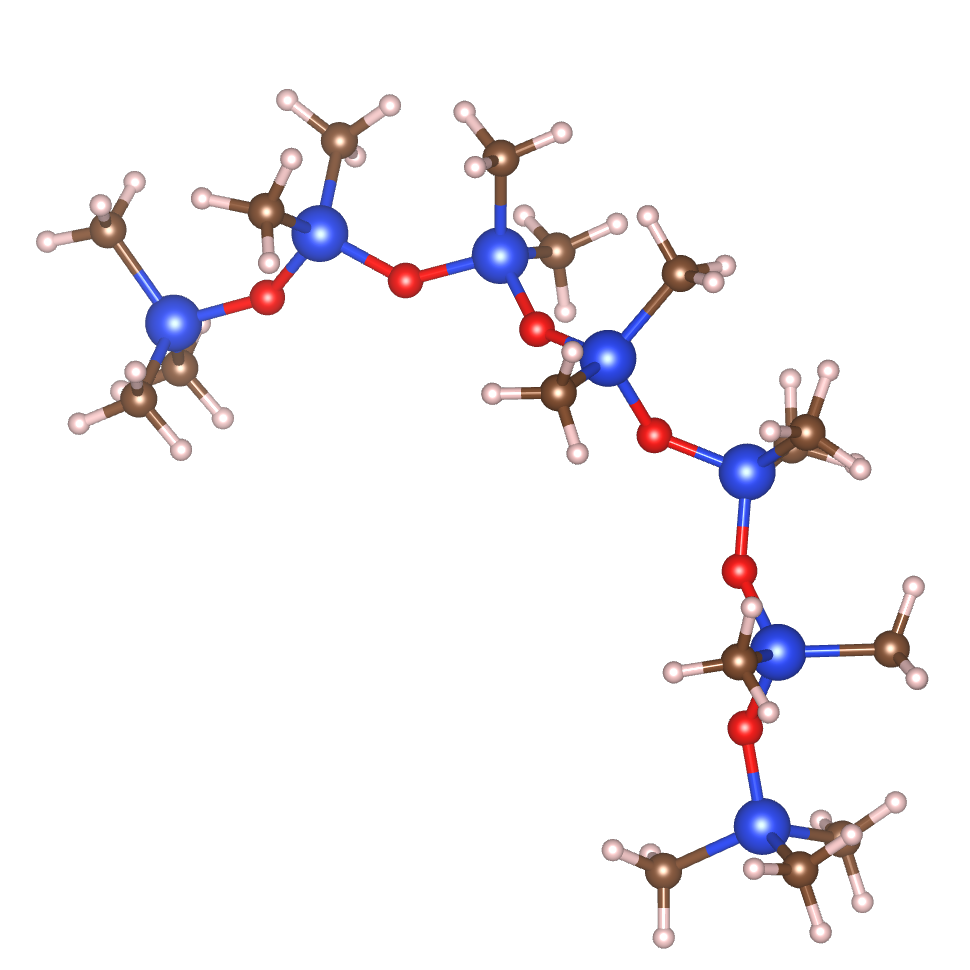
GRACE provides the accuracy needed to predict molecular conformations
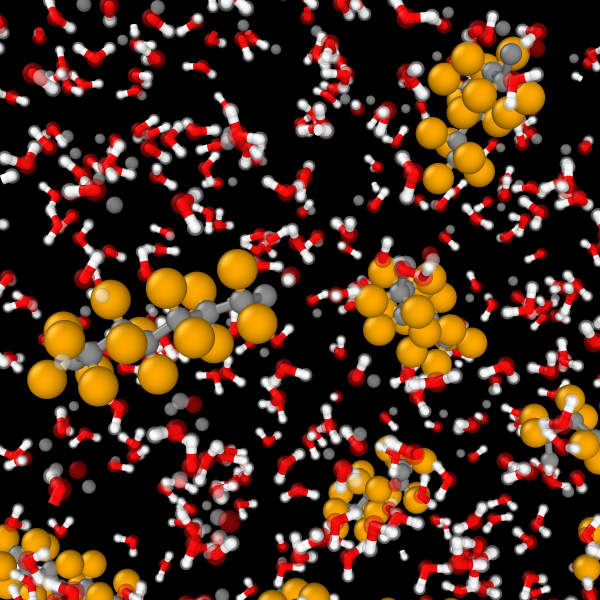
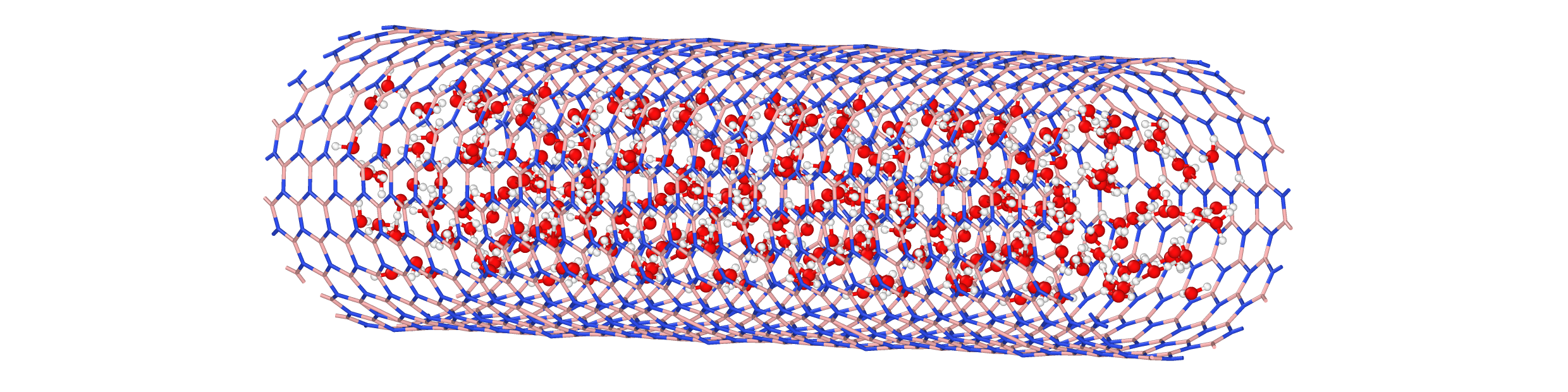
GRACE interatomic potentials provide the efficiency and accuracy needed for capturing solvents in solutions