Foundational models
GRACE foundational potentials represent a paradigm shift in computational materials science. Unlike traditional models limited to specific elements or alloys, GRACE utilizes a universal framework trained across the entire periodic table to predict atomic forces and energies with ab initio accuracy. This foundational approach allows researchers to bypass the computationally expensive constraints of density functional theory (DFT) while maintaining quantum-mechanical precision. By enabling simulations that are orders of magnitude faster than conventional methods, GRACE empowers scientists to explore vast chemical spaces and accelerate the discovery of novel materials without the need for system-specific model training.

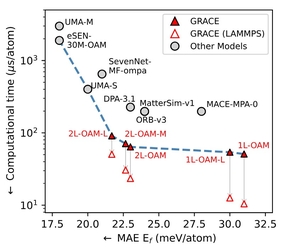
GRACE models establish a new Pareto front for accuracy versus efficiency among foundational interatomic potentials. They can be further adapted to specialized tasks and simpler architectures via fine-tuning and knowledge distillation, achieving high accuracy while preventing catastrophic forgetting.
Training datasets
GRACE foundational models have been trained on several extensive open-source DFT datasets including:
– OMat24 (Open Materials 2024)
Currently one of the largest publicly available datasets for materials property prediction, containing approximately 110 million DFT calculations covering 89 elements. It includes a high volume of non-equilibrium structures generated via Boltzmann sampling and ab initio molecular dynamics (AIMD). This diversity allows the model to learn forces and energies for structures far from equilibrium, which is critical for stable simulations.
– Alexandria (specifically the “sAlex” subsample)
The Alexandria library is a repository of 3D crystal structures and their properties. GRACE foundational models typically use a subsampled version (often cited as 10.4 million structures) to efficiently capture a wide variety of solid-state chemistries without redundant data processing.
– MPtrj (Materials Project Trajectory)
This dataset consists of full relaxation trajectories from the Materials Project. It contains roughly 1.6 million structures (associated with ~146,000 materials). Because it includes the “steps” taken during structural relaxation (not just the final result), it teaches the potential how atoms move toward their lowest energy states, which is essential for accurate geometry optimization.
– OMol25 (Open Molecules 2025)
This is the molecular counterpart to the OMat24 dataset. While OMat24 focuses on inorganic bulk materials (crystals), OMol focuses on finite molecular systems. It contains over 100 million DFT calculations covering approximately 83 million unique molecular systems, including large biomolecules (protein fragments, ligands), metal complexes (transition metals, organometallics), and electrolytes (solvated ions, battery components).